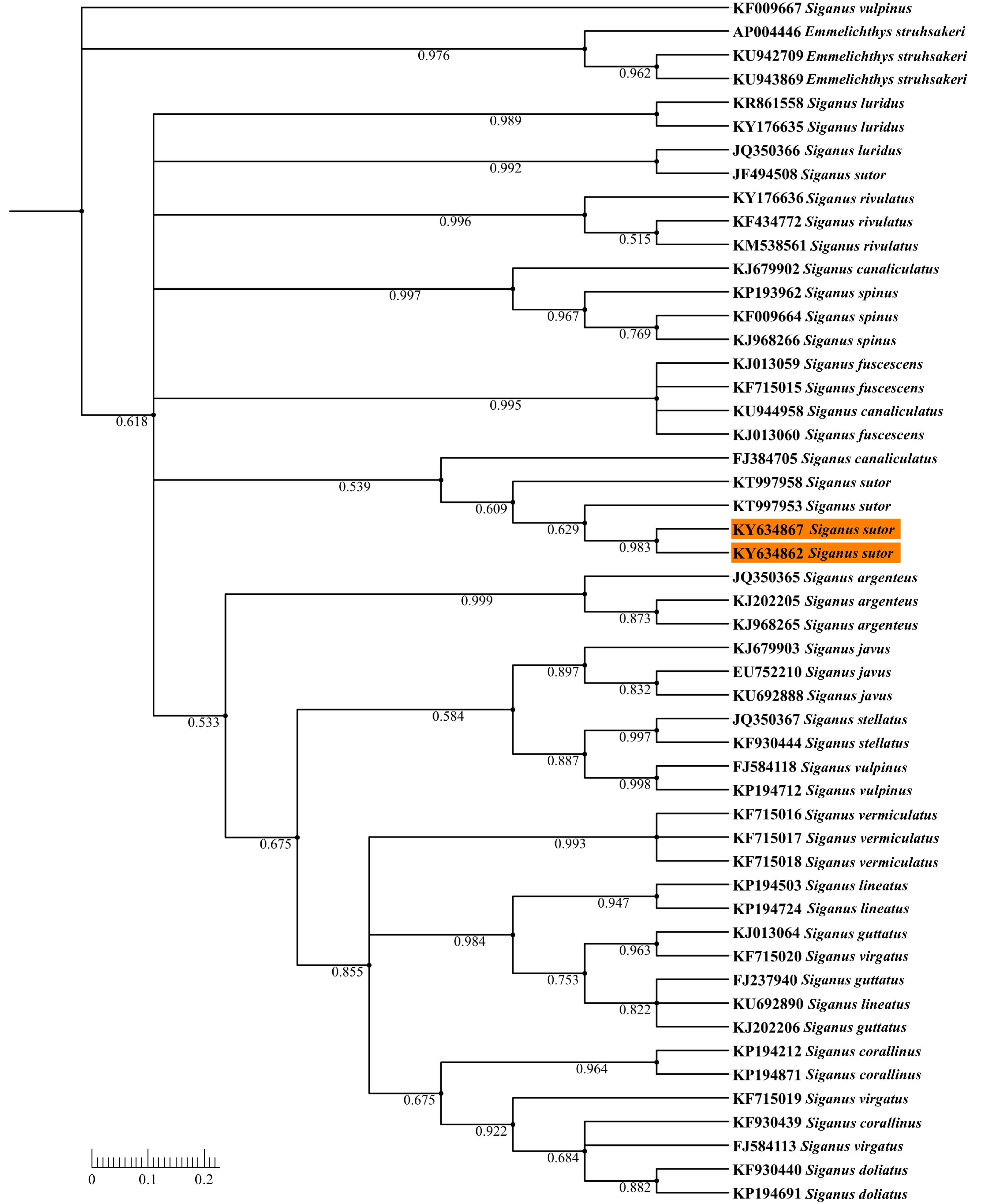
| Citation: | Tapan K. Barik, Surya N. Swain, Bijayalaxmi Sahu, Bibarani Tripathy, Usha R. Acharya. Morphological and molecular evidence supports the first occurrence of two fishes, Siganus sutor (Valenciennes, 1835) and Seriolina nigrofasciata (Rüppell, 1829) (Actinopterygii: Perciformes), from marine waters of Odisha coast, Bay of Bengal, India[J]. Acta Oceanologica Sinica, 2020, 39(6): 26-35. doi: 10.1007/s13131-020-1609-x

|
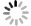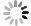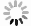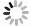
| [1] |
Allendorf F W, Hohenlohe P A, Luikart G. 2010. Genomics and the future of conservation genetics. Nature Reviews Genetics, 11(10): 697–709. doi: 10.1038/nrg2844
|
| [2] |
Altschul S F, Gish W, Miller W, et al. 1990. Basic local alignment search tool. Journal of Molecular Biology, 215(3): 403–410. doi: 10.1016/S0022-2836(05)80360-2
|
| [3] |
Arvedlund M. 2009. First records of unusual marine fish distributions-can they predict climate changes?. Journal of the Marine Biological Association of the United Kingdom, 89(4): 863–866. doi: 10.1017/S002531540900037
|
| [4] |
Bandelt H J, Forster P, Röhl A. 1999. Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution, 16(1): 37–48. doi: 10.1093/oxfordjournals.molbev.a026036
|
| [5] |
Barik T K, Swain S N, Sahu B, et al. 2018. Morphological and genetic analyses of the first record of longrakered trevally, Ulua mentalis (Perciformes: Carangidae) and of the pinjalo snapper, Pinjalo pinjalo (Perciformes: Lutjanidae) in the Odisha coast, Bay of Bengal. Mitochondrial DNA Part A, DNA Mapping, Sequencing, and Analysis, 29(4): 552–560. doi: 10.1080/24701394.2017.1320993
|
| [6] |
Capella-Gutiérrez S, Silla-Martínez J M, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics, 25(15): 1972–1973. doi: 10.1093/bioinformatics/btp348
|
| [7] |
Clarke L M, Munch S B, Thorrold S R, et al. 2010. High connectivity among locally adapted populations of a marine fish (Menidia menidia). Ecology, 91(12): 3526–3537. doi: 10.1890/09-0548.1
|
| [8] |
Cowen R K, Paris C B, Srinivasan A. 2006. Scaling of connectivity in marine populations. Science, 311(5760): 522–527. doi: 10.1126/science.1122039
|
| [9] |
Davis M B, Shaw R G, Etterson J R. 2005. Evolutionary responses to changing climate. Ecology, 86(7): 1704–1714. doi: 10.1890/03-0788
|
| [10] |
DeWoody J A, Avise J C. 2000. Microsatellite variation in marine, freshwater and anadromous fishes compared with other animals. Journal of Fish Biology, 56(3): 461–473. doi: 10.1111/j.1095-8649.2000.tb00748.x
|
| [11] |
Eschmeyer W N, Fong J D. 2017. Species by Family/Subfamily. http://researcharchive.calacademy.org/research/ichthyology/catalog/SpeciesByFamily.asp [2017-04-28/2018-01-17]
|
| [12] |
Eschmeyer W N, Fricke R, van der Laan R. 2017. Catalog of Fishes: Genera, Species, References. http://researcharchive.calacademy.org/research/ichthyology/catalog/fishcatmain.asp [2017-04-28/2018-01-17]
|
| [13] |
Folmer O, Black M, Hoeh W, et al. 1994. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology, 3(5): 294–299
|
| [14] |
Froese R, Pauly D. 2017. FishBase. World Wide Web electronic publication. www.fishbase.org [2017-02/2018-02-10]
|
| [15] |
Gavrilets S. 2003. Perspective: models of speciation: what have we learned in 40 years?. Evolution, 57(10): 2197–2215. doi: 10.1111/j.0014-3820.2003.tb00233.x
|
| [16] |
Hajibabaei M, Singer G A C, Hebert P D N, et al. 2007. DNA barcoding: how it complements taxonomy, molecular phylogenetics and population genetics. Trends in Genetics, 23(4): 167–172. doi: 10.1016/j.tig.2007.02.001
|
| [17] |
Harley C D G, Hughes R A, Hultgren K M, et al. 2006. The impacts of climate change in coastal marine systems. Ecology Letters, 9(2): 228–241. doi: 10.1111/j.1461-0248.2005.00871.x
|
| [18] |
Hebert P D N, Cywinska A, Ball S L, et al. 2003. Biological identifications through DNA barcodes. Proceedings of the Royal Society B: Biological Sciences, 270(1512): 313–321. doi: 10.1098/rspb.2002.2218
|
| [19] |
Hey J, Nielsen R. 2007. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proceedings of the National Academy of Sciences of the United States of America, 104(8): 2785–2790. doi: 10.1073/pnas.0611164104
|
| [20] |
Hey J. 2010. Isolation with migration models for more than two populations. Molecular Biology and Evolution, 27(4): 905–920. doi: 10.1093/molbev/msp296
|
| [21] |
Hoffmann A A, Hallas R J, Dean J A, et al. 2003. Low potential for climatic stress adaptation in a rainforest Drosophila species. Science, 301(5629): 100–102. doi: 10.1126/science.1084296
|
| [22] |
Ivanova N V, Zemlak T S, Hanner R H, et al. 2007. Universal primer cocktails for fish DNA barcoding. Molecular Ecology Notes, 7(4): 544–548. doi: 10.1111/j.1471-8286.2007.01748.x
|
| [23] |
Johnson G, Gill A. 1998. Perches and their allies. In: Paxton J R, Eschmeyer W, eds. Encyclopedia of Fishes. 2nd ed. San Diego, CA: Academic Press
|
| [24] |
Kaltz O, Shykoff J. 1998. Local adaptation in host-parasite systems. Heredity, 81(4): 361–370. doi: 10.1046/j.1365-2540.1998.00435.x
|
| [25] |
Katoh K, Standley D M. 2016. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics, 32(13): 1933–1942. doi: 10.1093/bioinformatics/btw108
|
| [26] |
Kelly R P, Oliver T A, Sivasundar A, et al. 2010. A method for detecting population genetic structure in diverse, high gene-flow species. Journal of Heredity, 101(4): 423–436. doi: 10.1093/jhered/esq022
|
| [27] |
Kimura M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution, 16(2): 111–120. doi: 10.1007/BF01731581
|
| [28] |
Kirkpatrick M, Barton N H. 1997. Evolution of a species range. The American Naturalist, 150(1): 1–23. doi: 10.1086/286054
|
| [29] |
Levene H. 1953. Genetic equilibrium when more than one ecological niche is available. The American Naturalist, 87(836): 331–333. doi: 10.1086/281792
|
| [30] |
Limborg M T, Helyar S J, De Bruyn M, et al. 2012. Environmental selection on transcriptomederived SNPs in a high gene flow marine fish, the Atlantic herring (Clupea harengus). Molecular Ecology, 21(15): 3686–3703. doi: 10.1111/j.1365-294X.2012.05639.x
|
| [31] |
Marko P B, Hart M W. 2011. The complex analytical landscape of gene flow inference. Trends in Ecology & Evolution, 26: 448–456. doi: 10.1016/j.tree.2011.05.007
|
| [32] |
McCarty J P. 2001. Ecological consequences of recent climate change. Conservation Biology, 15(2): 320–331. doi: 10.1046/j.1523-1739.2001.015002320.x
|
| [33] |
Messing J. 1983. New M13 vectors for cloning. Methods in Enzymology, 101: 20–78. doi: 10.1016/0076-6879(83)01005-8
|
| [34] |
Milano I, Babbucci M, Cariani A, et al. 2014. Outlier SNP markers reveal fine-scale genetic structuring across European hake populations (Merluccius merluccius). Molecular Ecology, 23(1): 118–135. doi: 10.1111/mec.12568
|
| [35] |
Nielsen E E, Hemmer-Hansen J, Poulsen N A, et al. 2009. Genomic signatures of local directional selection in a high gene flow marine organism; the Atlantic cod (Gadus morhua). BMC Evolutionary Biology, 9: 276. doi: 10.1186/1471-2148-9-276
|
| [36] |
Ntiba M J, Jaccarini V. 1992. The effect of oocytic atresia on fecundity estimates of the rabbit fish Siganus sutor (Pisces: Siganidae) of Kenyan marine inshore waters. Hydrobiologia, 247(1–3): 215–222. doi: 10.1007/BF00008221
|
| [37] |
Palumbi S R. 2003. Population genetics, demographic connectivity, and the design of marine reserves. Ecological Applications, 13(sp1): 146–158. doi: 10.1890/1051-0761(2003)013[0146:PGDCAT]2.0.CO;2
|
| [38] |
Palumbi S R, Warner R R. 2003. Why gobies are like Hobbits. Science, 299(5603): 51–52. doi: 10.1126/science.1080775
|
| [39] |
Paxton J R, Hoese D F, Allen G R, et al. 1989. Pisces. Petromyzontidae to Carangidae. Zoological Catalogue of Australia, Vol. 7. Canberra: Australian Government Publishing Service, 1–665
|
| [40] |
Phillips S J, Anderson R P, Schapire R E. 2006. Maximum entropy modeling of species geographic distributions. Ecological Modelling, 190(3–4): 231–259. doi: 10.1016/j.ecolmodel.2005.03.026
|
| [41] |
Posada D, Crandall K A. 1998. Modeltest: testing the model of DNA substitution. Bioinformatics, 14(9): 817–818. doi: 10.1093/bioinformatics/14.9.817
|
| [42] |
Ronquist F, Huelsenbeck J P. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics, 19(12): 1572–1574. doi: 10.1093/bioinformatics/btg180
|
| [43] |
Sambrook J, Russell R W. 2001. Molecular Cloning: A Laboratory Manual, 3rd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press
|
| [44] |
Schliep K P. 2011. Phangorn: phylogenetic analysis in R. Bioinformatics, 27(4): 592–593. doi: 10.1093/bioinformatics/btq706
|
| [45] |
Shen Yanjun, Guan Lihong, Wang Dengqiang, et al. 2016. DNA barcoding and evaluation of genetic diversity in Cyprinidae fish in the midstream of the Yangtze River. Ecology and Evolution, 6(9): 2702–2713. doi: 10.1002/ece3.2060
|
| [46] |
Smith M M. 1986. Siganidae. In: Smith M M, Heemstra P C, eds. Smiths’ Sea Fishes. Berlin: Springer-Verlag, 824–825
|
| [47] |
Spicer G S, Gaston K J. 1999. Physiological Diversity and Its Ecological Implications. Oxford, UK: Blackwell Science Ltd
|
| [48] |
Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22(21): 2688–2690. doi: 10.1093/bioinformatics/btl446
|
| [49] |
Stewart J R, Lister A M. 2001. Cryptic northern refugia and the origins of the modern biota. Trends in Ecology & Evolution, 16(11): 608–613
|
| [50] |
Talwar P K, Kacker R K. 1984. Commercial sea fishes of India. Calcutta: Zoological Survey of India, 997
|
| [51] |
Tepolt C K. 2015. Adaptation in marine invasion: a genetic perspective. Biological Invasions, 17(3): 887–903. doi: 10.1007/s10530-014-0825-8
|
| [52] |
Therkildsen N O, Hemmer-Hansen J, Als T D, et al. 2013. Microevolution in time and space: SNP analysis of historical DNA reveals dynamic signatures of selection in Atlantic cod. Molecular Ecology, 22(9): 2424–2440. doi: 10.1111/mec.12260
|
| [53] |
Waples R S, Gaggiotti O. 2006. What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Molecular Ecology, 15(6): 1419–1439. doi: 10.1111/j.1365-294X.2006.02890.x
|
| [54] |
Woodland D J. 1990. Revision of the fish family Siganidae with descriptions of two new species and comments on distribution and biology. Indo-Pacific Fishes, 19: 136
|
| [55] |
Yang Ziheng. 1994. Estimating the pattern of nucleotide substitution. Journal of Molecular Evolution, 39(1): 105–111. doi: 10.1007/bf00178256
|

Figures(5) / Tables(2)

Supported by:
Beijing Renhe Information Technology Co. Ltd





 DownLoad:
DownLoad: