

Integrated transcriptome analysis of mRNA and miRNA revealed defensive strategies in the large yellow croaker Larimichthys crocea against Pseudomonas plecoglossicida infection
-
Abstract: Large yellow croaker (Larimichthys crocea) is an economically important fish, with the annual production ranking second among maricultured fish in China. Outbreaks of visceral white nodules disease caused by Pseudomonas plecoglossicida have led to substantial economic losses for the L. crocea aquaculture industry. However, L. crocea defense strategies against P. plecoglossicida infection, especially the role of microRNAs (miRNAs) in the defense against P. plecoglossicida, are poorly understood. Here, we analyzed changes in the mRNA and miRNA expression profiles in the spleen of L. crocea at 96 h post-infection and explored its defensive strategies. Principal component analysis (PCA) showed that P. plecoglossicida infection brought about a profound remodeling of both the miRNA and mRNA profiles. Enrichment analysis showed that the inflammatory response (IL-17 signaling pathway, chemokines and chemokine receptor pathway), ATP synthesis (TCA cycle and oxidative phosphorylation), apoptosis and necroptosis (TNF signaling pathway), and proteolysis (proteasome pathway) were enriched and upregulated by P. plecoglossicida. Thus, P. plecoglossicida infection activated the inflammatory response, stimulated ATP synthesis, and accelerated apoptosis and necroptosis, and promoted proteasome-mediated protein degradation. Additionally, integrated analysis identified 568 miRNA–mRNA pairs. KEGG enrichment analysis of the miRNA targets showed that the enriched pathways included cytokine–cytokine receptor interaction, the chemokine signaling pathway, the C-type lectin receptor signaling pathway, and apoptosis. Integrated analysis identified 14 miRNAs which targeted 44 immune-related genes. Altogether, our results revealed not only the role of the inflammatory response, energy metabolism, apoptosis and necroptosis, and the proteasome pathway in L. crocea defense against P. plecoglossicida infection, but also the regulatory networks of miRNAs associated with host defense against P. plecoglossicida.
-
1. Introduction
Large yellow croaker (Larimichthys crocea), a species of Sciaenidae in Perciformes, is an economically important marine fish, with the annual production ranking second among mariculture fish species in China (Ministry of Agriculture and Rural Affairs et al., 2024). The L. crocea industry suffers substantial economic losses from visceral white nodules disease (VWND), which is caused by P. plecoglossicida infection (Zhang et al., 2014; Li et al., 2020c). Pseudomonas plecoglossicida is a temperature-sensitive Gram-negative bacterium and usually results in the outbreak of VWND at a water temperature of 15−24℃ in several cultured fish (Nishimori et al., 2000; Akayli et al., 2011; Huang et al., 2018a, 2019; Zhang et al., 2018). Recently, we isolated a strain named PQLYC4, which resulted in VWND at 12℃ (Li et al., 2020c), a much lower temperature than reported previously. The whole genome of several strains of P. plecoglossicida has been sequenced, and multiple potential virulence genes were identified (Mao et al., 2013; Huang et al., 2018b). The function of many virulence genes, especially those for secretion systems and flagellar synthesis, has been well documented. For example, FLIA is a gene that regulates flagellar synthesis in pathogenic bacteria. Infection of the fliA-RNAi strain increased the survival rate of L. crocea by 30% compared with those infected by the wild type. Comparative transcriptome analysis of L. crocea spleen showed that fliA-RNAi strain infection decreased the expression of some proinflammatory genes, including IL-1β and TNFα, compared with infection by the wild-type strain (Sun et al., 2019a). Similar methods have been used to elucidate the function of other virulent genes, such as secY (Wang et al., 2019) (a component of Sec protein secretion system), impB (Liu et al., 2020) (one core component of the type VI secretion system), ClpV (Luo et al., 2019) (a gene for T6SS), flgM (Sun et al., 2019b) (which regulates flagellar synthesis), flgK (Bulieris et al., 2017) (which is involved in flagellar assembly), and fliG (Jiao et al., 2021) (a rotor protein of the bacterial flagellar) in P. plecoglossicida. However, the defensive mechanisms of L. crocea against P. plecoglossicida infection are still poorly understood.
MicroRNAs (miRNAs) are small non-coding RNAs (Zhou et al., 2021), which mediate post-transcriptional regulation of gene expression. miRNAs mainly inhibit gene expression by repressing translation or promoting mRNA degradation (Fu et al., 2020). miRNAs are widely engaged in the host immune response in fishes (Zhou et al., 2021). For example, pol-miR-novel_171 plays a vital role in apoptosis and anti-bacterial immunity by targeting the FAM49B gene (Li et al., 2020a). miR-146a can regulate apoptosis and NF-κB, which further promotes Singapore grouper iridovirus infection (Ni et al., 2017). Pol-miR-novel_547 is involved in the pathogen infection, autophagy, and apoptosis in Japanese flounder by target the PTEN gene (Li et al., 2020b). However, it is still poorly understood that how miRNAs counteract the P. plecoglossicida infection in L. crocea. Recently, we characterized the mRNA and miRNA profiles in L. crocea spleen infected by P. plecoglossicida at 12 h and 24 h post-infection, and revealed the microRNA‐mediated immune response at early stage (Chen et al., 2023). Here, we characterized the changes of mRNAs and miRNAs profile after P. plecoglossicida infection by comparative transcriptome analysis at 96 h post-infection, found that inflammatory response (IL-17 signaling pathways, chemokines and chemokine receptor pathway), ATP synthesis, apoptosis and necroptosis, and proteasome-mediated protein degradation were activated. Furthermore, the regulatory mechanism of miRNAs in resistance to P. plecoglossicida infection was also clarified. Our results elucidated the miRNA-mediated defensive strategy of L. crocea against P. plecoglossicida at the late stage of infection.
2. Materials and methods
2.1 Bacterial strain and culture conditions
Pseudomonas plecoglossicida PQLYC4, a strain previously isolated from L. crocea suffering from VWND at a water temperature of 12℃, was used for experimental infection (Li et al., 2020c). The culture conditions referred to our previous reports (Zhang et al., 2021a).
2.2 Experimental infection and sampling
The experimental infection and sampling were performed as previously described (Zhang et al., 2021a). Briefly, 16 L. crocea (100±20 g), bought from Ningde Fufa Company in Ningde City, Fujian Province, China, were kept in one ton of aerated seawater at 18℃ for 2 weeks. Pseudomonas plecoglossicida was diluted with 1×PBS to a concentration of 1×103 colony-forming units (CFU) per milliliter. Then, 16 fish were divided randomly into two groups with eight individuals in each group. The experimental group was injected intraperitoneally 200 μL of 1×103 CFU/ml of P. plecoglossicida suspension. The control group was injected intraperitoneally 200 μL of sterile PBS. All the individuals were anaesthetized using MS222 at 96 h after infection. Their spleens were harvested and snap-frozen in liquid nitrogen. Six samples, of which three from injected group and the other three from control group, were sent to Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai City, China) for RNA-seq. Ten samples, harvested from the injected and control groups, respectively, were used for quantitative real-time PCR (qRT-PCR). All the animal experiments were carried out in strict accordance with the regulations of the Administration of Affairs Concerning experimental Animals approved by the Animal Care and Use Committee of Fujian Agriculture and Forestry University.
2.3 Transcriptome sequencing
Eastep Super Total RNA Extraction Kit (Promega) was used to extract the total RNA from the spleen. The purity and concentration of RNA were determined by Nanodrop2000. Agarose gel electrophoresis and RNA integrity number (RIN) determined by Agilent2100 were used to assess the RNA integrity. Then mRNA and small RNA (sRNA) libraries were prepared, following the previous report (Ou et al., 2021). Briefly, for mRNA sequencing, mRNAs were isolated by using oligo (dT) magnetic beads, and broken into 300 bp approximately fragments in a fragmentation buffer subsequently. Fragmented mRNA was reverse transcribed into the first-strand cDNAs using random hexamers as primers, followed by the synthesis of the double-stranded cDNAs. Sequencing adaptors were ligated to the double-stranded cDNAs, which were further enriched by 15 cycles of PCR amplification. Proper DNA fragments were recovered by agarose gel electrophoresis and used for paired-end 150 bp sequencing on Illumina Novaseq 6000. For sRNA sequencing, sRNA library was prepared by TruSeq small RNA library preparation kits (Illumina). The library was also sequenced on the Illumina Novaseq 6000. All the raw data in this study has been deposited in sequence read archive (SRA) database with accession number SRR23279742-SRR23279747 for mRNA and SRR23250140-SRR23250145 for miRNA.
2.4 Evaluation of RNA-seq quality
Clean reads produced from mRNA-seq and sRNA-seq were aligned against the L. crocea genome by using HISAT2 (Kim et al., 2019) and bowtie (Langmead et al., 2009) with default parameters to compute the mapping rate. Considering that mRNA-seq with unsaturated sequencing depth can lead to imprecise estimations of the mRNA abundance, the sample that has the least clean reads was used for sequencing saturation analysis by RSeQC (Version 2.3.6) (Wang et al., 2012). Additionally, principal component analysis (PCA) was performed to visualize the distances between samples (Son et al., 2018) by using the sklearn package (Rosenblatt et al., 2021).
2.5 Identification of DEGs
Adaptor sequences and low-quality reads were removed to obtain clean reads. HISAT2 (Kim et al., 2019) and StringTie (Version 1.3.3b) (Pertea et al., 2015) were respectively used to align the clean reads against the L. crocea genome and assemble the clean reads into transcripts. Expression abundance was quantified by RSEM (Version 1.3.1) (Li and Dewey, 2011), and expressed as transcripts per million reads (TPM). Differential expression analysis was carried out by DESeq2 (Version 1.24.0) (Love et al., 2014). The DEGs or differentially expressed mRNAs (DEMs) were defined as genes with |log 2fold changes (FC)| ≥ 1 and false discovery rate (FDR) <0.05.
2.6 Enrichment analysis based on the DEGs and GSEA
DEG-based GO and KEGG enrichment analysis were performed to identify the main function of the DEGs, following previous description (Zhang et al., 2021a). Briefly, Goatools (Version 0.6.5) (Klopfenstein et al., 2018) and a R script were respectively employed to perform GO and KEGG enrichment analysis with Fisher's exact test (Kanehisa et al., 2017). The KEGG pathways or GO terms with FDR<0.05 were regarded as significantly enriched pathways (SEPs) or significantly enriched GO terms (SEGTs). Additionally, gene set enrichment analysis (GSEA) (Mootha et al., 2003; Powers et al., 2018) was applied to determine the coordination of gene expression, taking each of the GO terms or KEGG pathways as one gene set. The GO terms or KEGG pathways with FDR<0.05 were considered as SEGTs or SEPs.
2.7 Identification of miRNAs and differentially expressed miRNAs (DEMIs)
Raw reads were cleaned by trimming adaptor sequence, and filtering reads in low quality or reads with a length greater than 32 nr or less than18 nr. Then, the clean reads were aligned to L. crocea genome, and the mapped reads were further used to blast against the miRbase (Griffiths-Jones et al., 2006) and Rfam (Kalvari et al., 2018, 2021) database to obtain known miRNAs and non-coding RNA (ncRNA), respectively. The unmapped reads were re-aligned against the reference genome and used for novel miRNA prediction by miRDeep2 (Friedländer et al., 2012). The miRNA expression level was expressed as transcripts per million (TPM) calculated by using the quantifier program in the miRDeep2. miRNAs with fold change not less than 2 and p-value less than 0.05 were viewed as DEMIs, which were identified by DESeq2.
2.8 miRNA target prediction and function annotation
The methods for the prediction of miRNA targets refer to previous reports (Tian et al., 2018; Liu et al., 2021). Briefly, TargetScan (Lewis et al., 2005), miranda (Lewis et al., 2005) and RNAhybrid (Krüger and Rehmsmeier, 2006) were used to predict miRNA-mRNA pairs based on the DEMIs and DEMs. For Targetscan, we excluded the type of 6mer and 7mer-1a, which have been suggested as lower efficacy (Riffo-Campos et al., 2016). For Miranda, only the miRNA-mRNA pairs with a free energy less than -20 kcal/mol were retained. For RNAhybrid, parameters were set as energy cutoff less than -10 kcal/mol and p-value cutoff less than 0.05. The potential miRNA-mRNA pairs must be supported by at least two software. Furthermore, the expression profile of miRNA and miRNA should show a negative correlation (r<-0.8 and p<0.05). GO and KEGG enrichment analysis were performed to determine the main function of the miRNA targets, and p<0.05 was used as the cutoff for significance. Furthermore, the potential interaction network of miRNA-mRNA was displayed by using Cytoscape V3.4 (Shannon et al., 2003) (http://cytoscape.org/).
2.9 qRT-PCR validation for miRNA and mRNA expression
To evaluate the repeatability of the RNA-seq, the relative expression level of 8 miRNAs and 9 mRNAs was validated by qRT-PCR. For mRNA, the methods for the first-strand cDNA synthesis and qRT-PCR follow our previous report (Zhang et al., 2021a). For miRNA, the first strand was synthetized by using the miRNA First Strand cDNA Synthesis (Tailing Reaction) kit (Sangon) according to its manual. qRT-PCR was performed in a 20 μL reaction mix containing 10 μL of qMix (Life Technologies), 0.2 μL of forward or reverse primer, 0.2 μL of cDNA temple. β-actin and U6 were used as the internal reference for the mRNA and miRNA quantification, respectively. The relative expression level of mRNA and miRNA was respectively normalized to β-actin and U6 by using the 2-ΔΔCt method, and expressed as fold changes of mRNA or miRNAs in the injected group relative to that in the control group. The primers used for miRNA and mRNA quantification were listed in Table S1. Graphpad Prism 8 was used to analyze the data, which were expressed as means ± standard deviation (SD). P<0.05 was used as the cutoff for significance.
3. Results
3.1 Assessment of the RNA-seq data
The transcriptome sequencing statistics for mRNA and sRNA were given in Table S2 and Table S3. mRNA-seq yielded 62.4-84.0 million raw reads, and 41.4-55.6 million clean reads were kept after removal of adaptor and low-quality reads. The Q20 of the clean reads ranges from 95.73% to 95.9%. About 82.4%-84.0% of the clean reads can be mapped to the L. crocea genome (Table S4). Saturation analysis showed that mRNA-seq produced sufficient read depth to accurately quantify the gene expression, even for the sample with the least clean reads (Fig. S1). A total of 10.1-12.8 million raw reads were generated from sRNA-seq, of which 8.9-11.0 million were produced following the read cleaning. The PCA plot of both the mRNA-seq and sRNA data clearly discriminated the injected group and the control group, indicating that P. plecoglossicida injection induced a conspicuous alteration of mRNA and miRNA profile (Fig. 1). For mRNA, the first principal component (PC1) contributed approximate 60.9% variance (Fig. 1a). For the miRNA data, the distances among the three samples in the injected group were much closer to each other than those among the three samples in the control group (Fig. 1b), which implied that the miRNA expression patterns may have become more similar in response to the P. plecoglossicida injection.
 Figure 1. Principal component analysis (PCA) depicts the relationship of the samples used for RNA-seq. a. PCA of the mRNA-seq data, and b. PCA of sRNA-seq data. PC1 and PC2 refer to the first and second principal component, respectively.
Figure 1. Principal component analysis (PCA) depicts the relationship of the samples used for RNA-seq. a. PCA of the mRNA-seq data, and b. PCA of sRNA-seq data. PC1 and PC2 refer to the first and second principal component, respectively.3.2 DEG identification and DEG-based enrichment analysis
A total of 4 452 genes were identified as DEGs, of which 2 161 were upregulated and 2 291 were downregulated in injected group (Fig. S2). GO enrichment analysis identified 45 SEGTs, including 22 subclasses of “biological process”, 6 subclasses of “cellular component” and 17 subclasses of “molecular function” (Fig. 2a). Ten GO terms were associated with the immunity, which included the top two enriched terms namely “immune response” and “immune system process” in the “biological process” category (Fig. 2a). Seven terms are related to proteasome or peptidase, among which “threonine-type endopeptidase activity” and “threonine-type peptidase activity” were the top two enriched terms in the “molecular function”, and the other five constituted the main part of the “cellular component” category (5/6) (Fig. 2a). Thus, P. plecoglossicida infection exerted a serious impact on the expression of genes related to the immune system, proteasome or peptidase.
 Figure 2. GO and KEGG Enrichment analysis of differentially expressed genes (DEGs). a. GO enrichment analysis of the DEGs. The GO terms associated with immunity, proteasome or peptidase were pointed by arrows in blue and green color, respectively. The x axis and y axis represent enrichment levels and GO terms, respectively. b. the top thirty significantly enriched pathways. The x axis and y axis refer to rich factor and pathways, respectively. Pathways involved in immune system, infectious disease, energy metabolism and proteasome were pointed by arrows in blue, yellow, red and green color respectively.
Figure 2. GO and KEGG Enrichment analysis of differentially expressed genes (DEGs). a. GO enrichment analysis of the DEGs. The GO terms associated with immunity, proteasome or peptidase were pointed by arrows in blue and green color, respectively. The x axis and y axis represent enrichment levels and GO terms, respectively. b. the top thirty significantly enriched pathways. The x axis and y axis refer to rich factor and pathways, respectively. Pathways involved in immune system, infectious disease, energy metabolism and proteasome were pointed by arrows in blue, yellow, red and green color respectively.KEGG enrichment analysis based on the DEGs also reached the same conclusion. A total of 63 pathways were identified as SEPs (Table S5). Of the top thirty SEPs, 18 belong to immune system and infectious disease (Fig. 2b). Additionally, proteasome was the most enriched pathway (Fig. 2b). Furthermore, all the DEGs in this pathway were upregulated in the injected group (Fig. S3). These suggested that immunity and proteasome in the L. crocea were significantly influenced by the P. plecoglossicida infection.
3.3 GSEA-based GO and KEGG enrichment analysis
The coordination of the gene expression was researched by GSEA-based GO and KEGG enrichment analysis. Both results illustrated that the main changes concentrated on the proteolysis, immunity and energy metabolism. A total of 38 GO terms were significantly enriched, of which 30 and 8 were upregulated in the injected group and the control group, respectively (Table S6). Among the 30 upregulated terms, 6 were for proteolysis, 4 for immune system or defense response, and 9 for ATP synthesis. The 6 proteolysis-related terms comprised “proteasome complex”, “threonine-type endopeptidase activity”, “proteasome core complex”, “proteolysis”, “proteasome-mediated ubiquitin-dependent protein catabolic process” and “proteolysis involved in cellular protein catabolic process” (Figs 3a-d and Table S6). Especially, upregulation occurred on all the genes encoding proteasome complex (Fig. 3a). The 4 terms related to immune system or defense response were respectively “defense response to gram-positive bacterium” and “chemokine activity”, “inflammatory response” and “immune response” (Figs 3e-f and Table S6). The 9 terms associated with ATP synthesis were “ATP hydrolysis coupled proton transport”, “respiratory chain”, “ATP synthesis coupled proton”, “mitochondrial respiratory chain”, “mitochondrial intermembrane space”, “mitochondrial intermembrane membrane”, “tricarboxylic acid cycle”, “cytochrome-c oxidase activity” and “mitochondrial respiratory chain complex Ⅰ” (Figs 3g-h and Table S6). Thus, the GSEA-based GO enrichment analysis demonstrated that proteolysis, ATP synthesis and immunity in L. crocea were activated by the P. plecoglossicida infection.
 Figure 3. GSEA enrichment plots of the significantly upregulated GO terms associated with proteolysis, immunity and ATP synthesis in the injected group. a-d. GSEA enrichment plots of the GO terms involved in the proteolysis, e-f. GSEA enrichment plots of the GO terms associated with immunity, and g-h. GSEA enrichment plots of the GO terms implicated in ATP synthesis. Each vertical line refers to one gene in the gene set. The lines in blue and red area strand for upregulated and downregulated genes in the injected group, respectively.
Figure 3. GSEA enrichment plots of the significantly upregulated GO terms associated with proteolysis, immunity and ATP synthesis in the injected group. a-d. GSEA enrichment plots of the GO terms involved in the proteolysis, e-f. GSEA enrichment plots of the GO terms associated with immunity, and g-h. GSEA enrichment plots of the GO terms implicated in ATP synthesis. Each vertical line refers to one gene in the gene set. The lines in blue and red area strand for upregulated and downregulated genes in the injected group, respectively.Similarly, GSEA-based KEGG enrichment analysis showed the upregulated pathways focus on the proteasome, energy metabolism and immunity. A total of 42 pathways were significantly enriched, of which 39 upregulated in the injected group and 3 upregulated in the control group (Table S7). Proteasome was the most significantly enriched pathway among all the 39 upregulated ones (Fig. 4a and Table S7), which is consistent with the DEGs-based KEGG analysis (Fig. 2b). “Oxidative phosphorylation” and “citrate cycle (TCA cycle)”, which are implicated in ATP synthesis, were upregulated in the injected group (Figs 4b-c and Table S7). Immune system, immune disease and infectious disease occupied the most part of the upregulated pathways (17/39) (Table S7). There are 7 pathways related to immune system, which were “IL-17 signaling pathway”, “toll-like receptor signaling pathway”, “cytosolic DNA-sensing pathway”, “c-type lectin receptor signaling pathway”, “complement and coagulation cascades”, “TNF signaling pathway” and “cytokine-cytokine receptor interaction” (Figs 4d-h and Table S7). Therefore, P. plecoglossicida infection accelerated proteasome-mediated protein degradation, stimulated the energy metabolism and activated the immune system in the L. crocea.
 Figure 4. GSEA enrichment plots of the significantly upregulated pathways associated with proteasome, ATP synthesis and immune system in injected group. a. GSEA enrichment plots of proteasome; b and c. GSEA enrichment plots of “TCA cycle” and “oxidative phosphorylation” pathways, both of which participate in energy metabolism; and d-h. GSEA enrichment plots of the pathways in immune system. Each vertical line refers to one gene in the gene set. The lines in blue and red area strand for upregulated and downregulated genes respectively.
Figure 4. GSEA enrichment plots of the significantly upregulated pathways associated with proteasome, ATP synthesis and immune system in injected group. a. GSEA enrichment plots of proteasome; b and c. GSEA enrichment plots of “TCA cycle” and “oxidative phosphorylation” pathways, both of which participate in energy metabolism; and d-h. GSEA enrichment plots of the pathways in immune system. Each vertical line refers to one gene in the gene set. The lines in blue and red area strand for upregulated and downregulated genes respectively.3.4 Annotation of DEGs involved in immune system, energy metabolism, proteasomes, apoptosis and necroptosis
To better understand the defense response, the DEGs associated with immune system, energy metabolism and proteasome were identified. Most of the DEGs associated with the innate system were upregulated, including IL-17 signaling pathway (IL17C, IL-17RA, IL-17RB, IL-17RC, HSP90A, HSP90B), chemokines and chemokine receptors (CXCR3, CCR9, CCR11, CXCL10, CXCL9, CCL20, CCL19), and complement and coagulation cascades pathway (C3, C4, C6, C7, C8A, MBL, FD, C5AR1) (Table S8). For proteasome, all the 40 DEGs encoding the proteasome components were upregulated in the P. plecoglossicida-injected L. crocea (Fig. S3 and Table S8). For ATP synthesis, almost all the DEGs in the oxidative phosphorylation pathway were upregulated (Fig. S4 and Table S8). Additionally, upregulated genes also predominated in TNF signaling pathway, especially for the genes associated with apoptosis (FADD, CASP3, CASP8, CASP7) and necroptosis (RIP3, MLKL and Drp1) (Fig. S5 and Table S8). Thus, P. plecoglossicida infection activated inflammatory response, proteasomes, energy metabolism, apoptosis and necroptosis in L. crocea.
3.5 Analysis of miRNAs and their target genes
A total of 553 miRNAs were obtained, including 294 known and 259 novel ones. Forty-one miRNAs were identified as DEMIs, of which 25 were upregulated and 16 were downregulated in injected group (Fig. S6 and Table S9). A total of 568 miRNA-mRNA interaction pairs were constructed, which formed between 24 DEmiRNAs and 537 DEmRNAs. KEGG enrichment analysis of miRNA targets showed that 28 pathways were enriched (Fig. 5a). Nine of the 28 enriched pathways were grouped into immune system (3), immune disease (1) or infectious disease (5) (Fig. 5a). The 3 pathways in immune system are comprised of “cytokine-cytokine receptor interaction”, “chemokine signaling pathway”, and “c-type lectin receptor signaling pathway”. Additionally, a network consisting of 49 immune-related miRNA-mRNA pairs, which were formed between 14 miRNAs and 44 genes, were constructed (Fig. 5b and Table S10). The target genes include many key immune-related genes, such as chemokine and chemokine receptors (CXCR5, CCL17 and CXCR3), interleukin receptor (IL1RL1, IL18RAP, IL18R1), lectin (MBL1 and MBL2). Thus, miRNAs may play an important role in the defense response.
 Figure 5. KEGG enrichment analysis of the miRNA targets and prediction of immune-related miRNA-mRNA pairs. a. KEGG enrichment analysis of the miRNA targets. Pathways involved in immune system, infectious and immune disease were indicated by arrows in different color. b. The immune-related miRNA-mRNA pairs. Diamond and ellipse represent miRNA and mRNA respectively.
Figure 5. KEGG enrichment analysis of the miRNA targets and prediction of immune-related miRNA-mRNA pairs. a. KEGG enrichment analysis of the miRNA targets. Pathways involved in immune system, infectious and immune disease were indicated by arrows in different color. b. The immune-related miRNA-mRNA pairs. Diamond and ellipse represent miRNA and mRNA respectively.3.6 qRT-PCR validation of DEGs and DEMIs expression
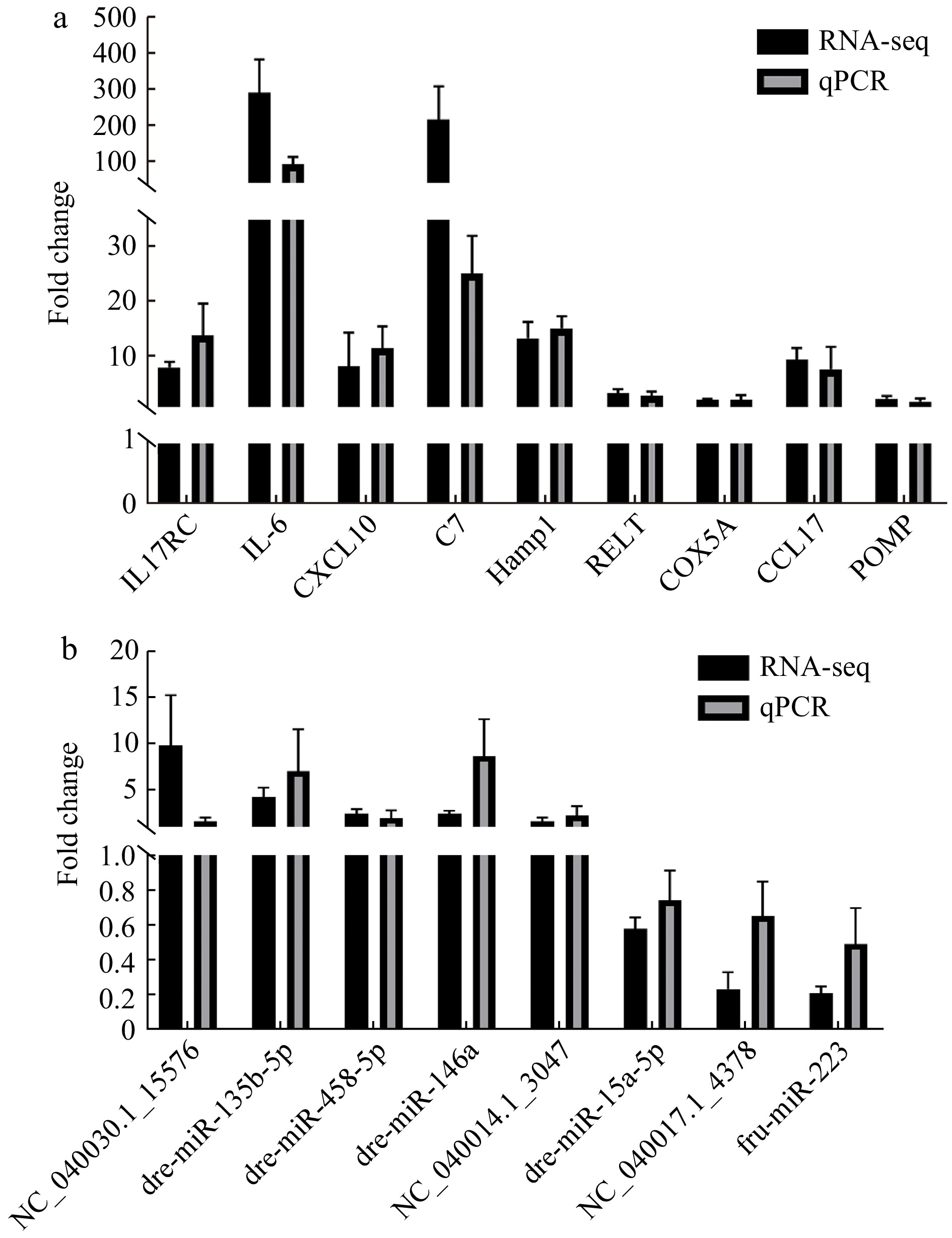
To assess the repeatability of RNA-seq, 9 DEGs and 8 DEMIs was selected and verified by using qRT-PCR (Fig. 6, Table S11). For DEGs, the expression of IL17-RC, IL-6, CXCL10, C7, Hamp1, RELT, COX5A, CCL17 and POMP were all upregulated in the L. crocea challenged by P. plecoglossicida (Fig. 6a, Table S11). For DEMIs, dre-miR-146a, NC_040030.1_15576, dre-miR-458-5p, NC_040014.1_3047, dre-miR-135b-5p were significantly upregulated, while fru-miR-223, NC_040017.1_4378 and dre-miR-15a-5p were significantly downregulated in the infected L. crocea (Fig. 6b, Table S11). The relative expression level of both DEGs and DEMIs from qRT-PCR were consistent with the those from RNA-seq data, illustrating the reliability of RNA-seq data.
 Figure 6. qRT-PCR verification of the 9 differentially expressed genes (DEGs) and 8 differentially expressed miRNAs (DEMIs). a. qRT-PCR verification of 9 DEGs, and b. qRT-PCR verification of 8 DEMIs.
Figure 6. qRT-PCR verification of the 9 differentially expressed genes (DEGs) and 8 differentially expressed miRNAs (DEMIs). a. qRT-PCR verification of 9 DEGs, and b. qRT-PCR verification of 8 DEMIs.4. Discussion
4.1 Pseudomonas plecoglossicida infection led to a profound transcriptome remodeling in L. crocea
Pseudomonas plecoglossicida infection has brought about great losses to the mariculture of L. crocea, a fish with annual output ranking second in China (Ministry of Agriculture and Rural Affairs et al., 2024). However, how L. crocea protects itself against P. plecoglossicida infection is still poorly understood. Here, we characterized the changes of mRNA and miRNA profile in the spleens of L. crocea infected by P. plecoglossicida at 96 h. The PCA plots clearly separated the control group and the infected group (Fig. 1), indicating that P. plecoglossicida infection has profoundly remodeled the miRNA and mRNA profile. Additionally, the expression of several DEGs and DEMIs was well validated by qRT-PCR, illustrating reliability and repeatability of the transcriptome data (Fig. 6).
4.2 Inflammatory response protects against P. plecoglossicida infection
Inflammatory response is essential in defense against pathogens. The inflammatory response in L. crocea was activated by the P. plecoglossicida infection. Significant enrichment and coordinately upregulated expression occurred on the IL-17 signaling pathways, chemokines and chemokine receptor (Figs 2 and 3 and Table S8). IL-17 signaling pathway is essential to ensure the elimination of extracellular pathogenic bacteria and inflammation (Qian et al., 2010). Chemokines mediate the recruitment of immune cells to the infected sites and enhance their cytotoxic function (Oo et al., 2010; Griffith et al., 2014; Khalil et al., 2021). Activation of the IL-17 signaling pathway and chemokines also occurred in many other pathogen-infected fishes (Dang et al., 2016; Maekawa et al., 2019), suggesting that the inflammatory response may be common in fishes.
4.3 Proteasome shapes the immune response against P. plecoglossicida infection
Proteasome extensively participates in the antiviral immune response in fishes. In sevenband grouper, proteasome subunit beta type-8 negatively regulates NF-κB responses during nervous necrosis viral infection (Krishnan et al., 2021). Proteasome in grass carp is involved in aquareovirus infection through the interaction between proteasome subunit beta-type 7 and grass carp reovirus (GCRV) capsid proteins (Wang et al., 2020). In Paralichthys olivaceus, proteasome was significantly enriched upon challenged by lymphocystis disease virus (Wu et al., 2018). However, the function of proteasome in bacterial infection received litter attention. In this study, the most striking change in the L. crocea spleen infected by P. plecoglossicida is the profoundly increased expression of genes encoding proteasome components. KEGG enrichment analysis based the DEGs showed that the proteasome is the most enriched pathway (Fig. 2b and Table S5). Furthermore, all the 40 DEGs encoding the proteasome components were upregulated by P. plecoglossicida infection (Fig. S3 and Table S8), which is consistent with the results of GSEA-based KEGG enrichment analysis (Fig. 4a). Especially, significant upregulation occurred on the PA28αβ (Table S8), which can promote the overall supply of MHC class I-binding peptides (de Graaf et al., 2011; Murata et al., 2018). The exact mechanism underlying the increased expression of genes for the proteasome components is still unknown, but considering the function of the proteasome in the MHC class I antigen processing (Kammerl and Meiners, 2016; Çetin et al., 2021), proteasome may mediate the initiation of the primary adaptive immune response against the P. plecoglossicida infection. This is supported by the fact that antigen processing and presentation pathway was significantly enriched (Fig. 2b). Additionally, proteasome also takes part in immune response by eliminating signaling components (Kammerl and Meiners, 2016; Çetin et al., 2021). Therefore, strengthen protein degradation by proteasome may contribute the generation of an adequate immune response against the P. plecoglossicida infection.
4.4 Pseudomonas plecoglossicida infection may activate the apoptosis and necroptosis mediated by TNF signaling pathway
Death of infected host cell is an intrinsic immune defense mechanism, which helps the clearance of intracellular pathogeny (Labbé and Saleh, 2008; Ashida et al., 2011; Behar and Briken, 2019). TNF is the most potent inducer of cell death among cytokines, which contributes cell death by either caspase-dependent way (apoptosis) or caspase-independent (necroptosis) (Tanzer, 2022). In this study, P. plecoglossicida infection significantly upregulated the expression of TNF and TNF receptor 1 (TNFR1) genes. KEGG enrichment analysis based on GSEA and DEGs showed that the TNF signaling pathway in L. crocea was significantly enriched, and most of the genes in the pathway were upregulated by P. plecoglossicida infection (Fig. 2b, 4f and S5). Furthermore, the expression of many apoptosis-induced genes including FADD, CASP8, CASP3 and CASP7 (Best, 2008; Kominami et al., 2012; McIlwain et al., 2013; Meng et al., 2021), and the necroptosis-induced genes including RIP3, MLKL and Drp1 (Nogusa et al., 2016; Li et al., 2021) significantly increased (Fig. S5 and Table S8). So, P. plecoglossicida infection activated the apoptosis and necroptosis mediated by TNF signaling pathway. Many studies suggest that the death of infected cells is generally beneficial for host. For example, macrophage apoptosis caused by Mycobacterium tuberculosis infection can increase host resistance with the help of efferocytosis (Martin et al., 2012; Behar and Briken, 2019). Some bacterial or viral pathogens can evade host defenses by inhibiting host apoptosis (Best, 2008; Behar and Briken, 2019). Necroptosis has also been considered as an important response against virus invasion (Nailwal and Chan, 2019). Thus, increased expression of the genes that favor cell death may provide a protective role against P. plecoglossicida infection.
4.5 Accelerated ATP synthesis provided energy basis for defense response
Another profound change in P. plecoglossicida-infected L. crocea is the enhanced ATP synthesis. GSEA-based GO enrichment analysis showed that “ATP hydrolysis coupled proton transport” and “respiratory chain” were significantly enriched, most genes in these GO terms were upregulated by the P. plecoglossicida infection (Figs 3g, h). Similarly, GSEA-based KEGG enrichment analysis also showed that upregulated genes predominated in the TCA cycle and oxidative phosphorylation pathways (Figs 4b, c). Furthermore, most of DEGs in these two pathways were upregulated (Table S8). Considering that the tremendously energetic requirement during defense response such as proteasome-mediated protein degradation, cell apoptosis, and immune-related gene expression, accelerated ATP synthesis may lay a foundation for the defense response for P. plecoglossicida infection.
4.6 miRNAs regulate the defense response against P. plecoglossicida infection through interaction with immune genes
miRNAs are endogenous small non-coding RNAs, which mainly negatively regulate gene expression (Baek et al., 2008; Andreassen and Høyheim, 2017; Zhang et al., 2021b). miRNAs widely participate in the immune response of fishes (Shen et al., 2019; Abo-Al-Ela, 2021; Sundaray et al., 2022). In this study, 568 DEmiRNA-DEmRNA pairs were predicted. KEGG enrichment analysis of the miRNA targets identified 9 immune-related pathways, including 3 in immune system (cytokine-cytokine receptor interaction, chemokine signaling pathway, and c-type lectin receptor), 6 infectious or immune disease (Fig. 5a). Interestingly, the c-type lectin receptor signaling pathway is also one of the most enriched pathways of the DEmiRNA target genes in the spleens of L. crocea infected by P. plecoglossicida at 24h, indicating that miRNAs may participate in the immune response through the c-type lectin receptor signaling pathway at the early and late stages of infection (Chen et al., 2023). Specifically, we constructed a network of 49 miRNA-mRNA pairs related to immune system (Fig. 5b), providing a foundation for further exploration the functions of miRNAs in immune defense. The miRNA targets included many genes for chemokine receptors, interleukin receptor and lectin (Table S10). Thus, miRNAs regulated the defense response against the P. plecoglossicida infection through interaction with immune-related genes.
-

Figure 1. Principal component analysis (PCA) depicts the relationship of the samples used for RNA-seq. a. PCA of the mRNA-seq data, and b. PCA of sRNA-seq data. PC1 and PC2 refer to the first and second principal component, respectively.
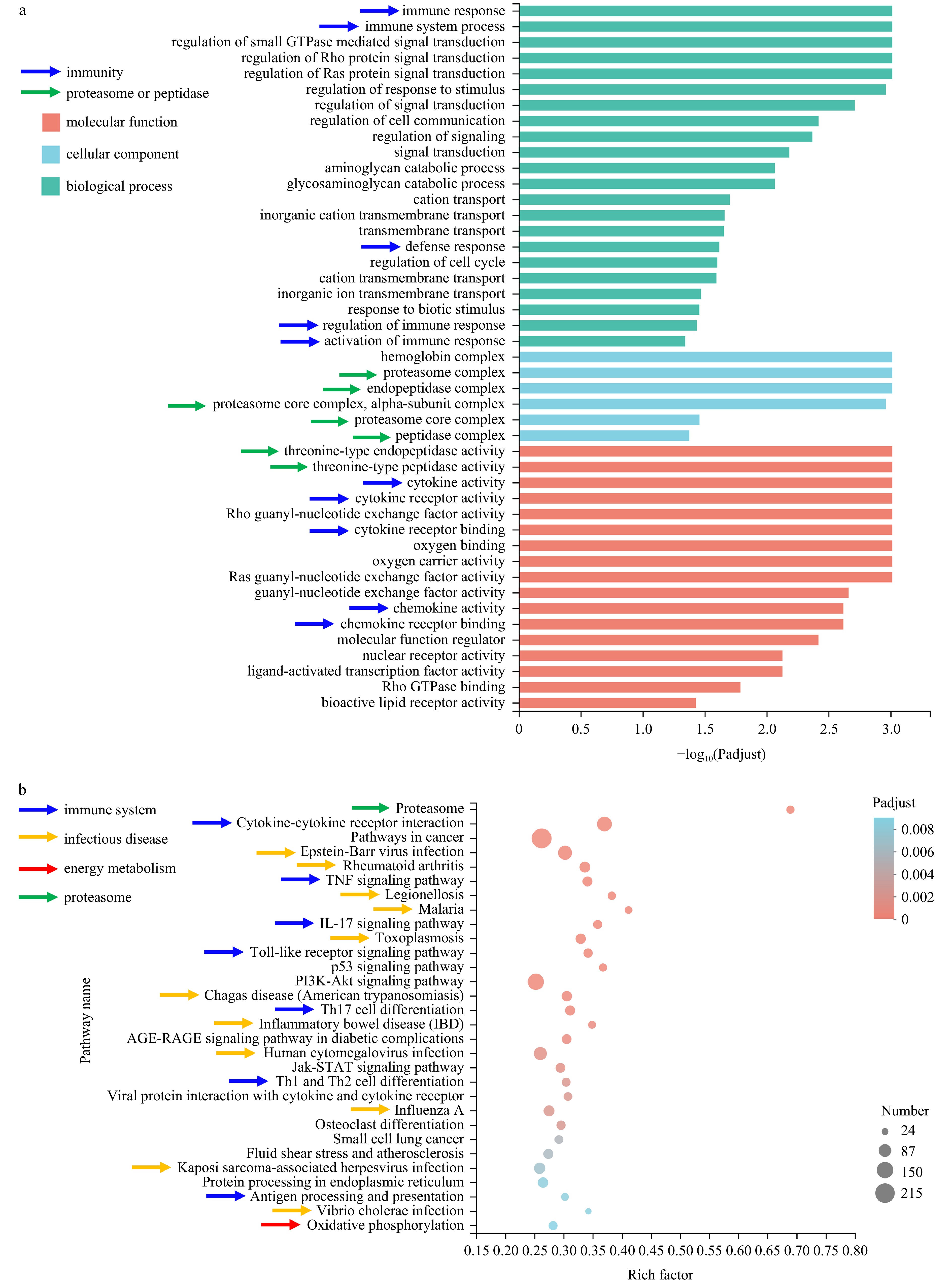
Figure 2. GO and KEGG Enrichment analysis of differentially expressed genes (DEGs). a. GO enrichment analysis of the DEGs. The GO terms associated with immunity, proteasome or peptidase were pointed by arrows in blue and green color, respectively. The x axis and y axis represent enrichment levels and GO terms, respectively. b. the top thirty significantly enriched pathways. The x axis and y axis refer to rich factor and pathways, respectively. Pathways involved in immune system, infectious disease, energy metabolism and proteasome were pointed by arrows in blue, yellow, red and green color respectively.
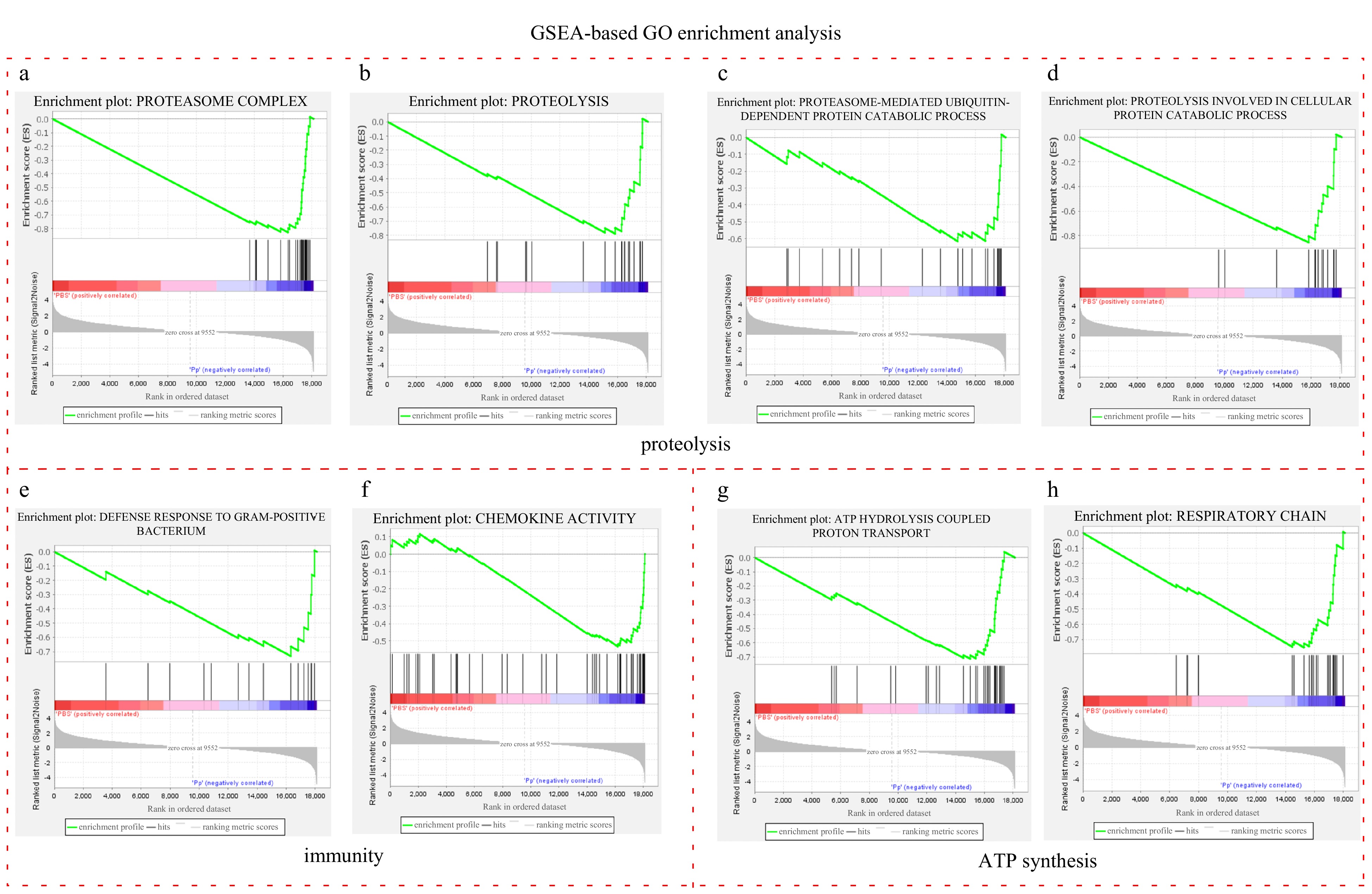
Figure 3. GSEA enrichment plots of the significantly upregulated GO terms associated with proteolysis, immunity and ATP synthesis in the injected group. a-d. GSEA enrichment plots of the GO terms involved in the proteolysis, e-f. GSEA enrichment plots of the GO terms associated with immunity, and g-h. GSEA enrichment plots of the GO terms implicated in ATP synthesis. Each vertical line refers to one gene in the gene set. The lines in blue and red area strand for upregulated and downregulated genes in the injected group, respectively.
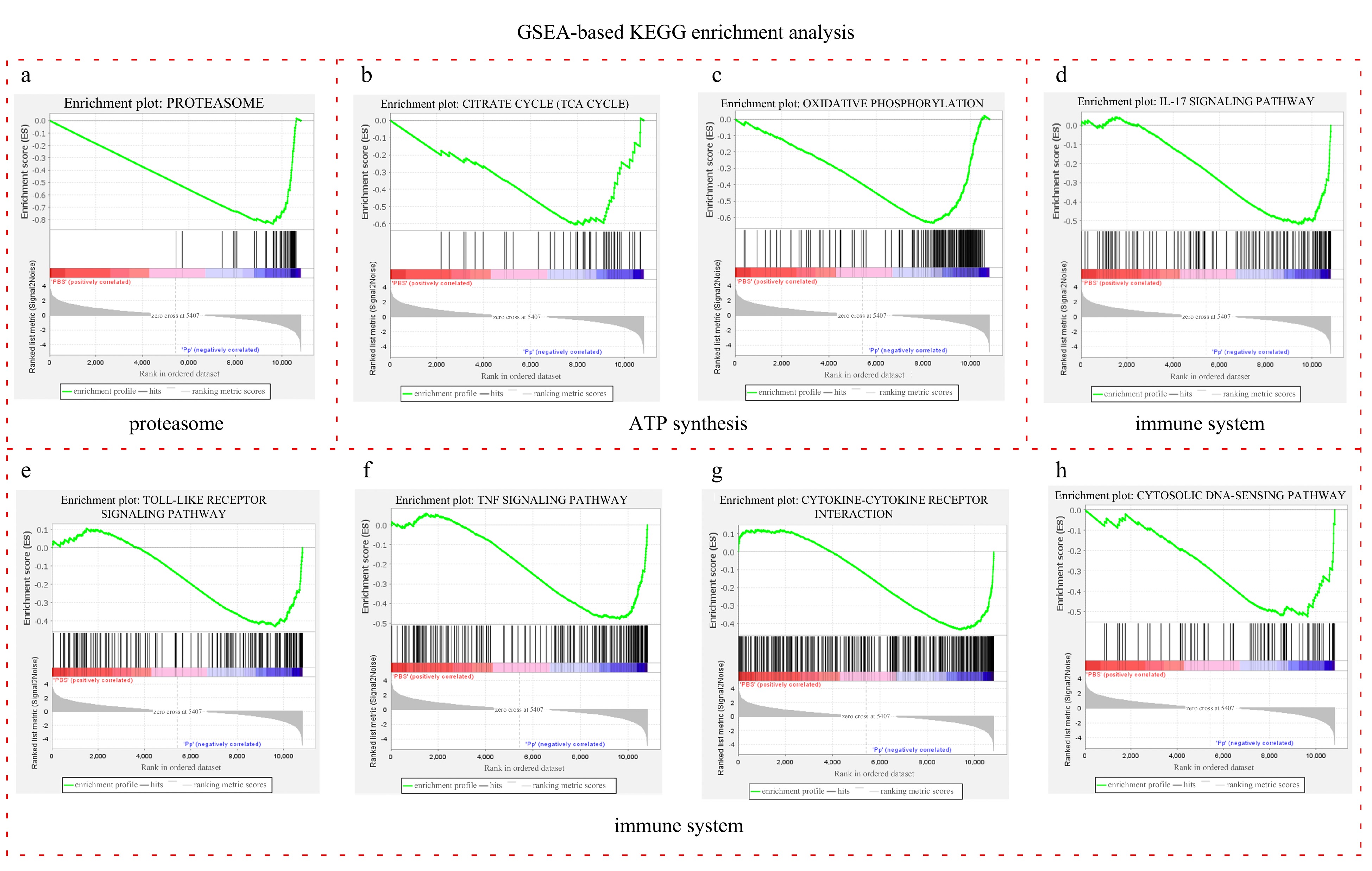
Figure 4. GSEA enrichment plots of the significantly upregulated pathways associated with proteasome, ATP synthesis and immune system in injected group. a. GSEA enrichment plots of proteasome; b and c. GSEA enrichment plots of “TCA cycle” and “oxidative phosphorylation” pathways, both of which participate in energy metabolism; and d-h. GSEA enrichment plots of the pathways in immune system. Each vertical line refers to one gene in the gene set. The lines in blue and red area strand for upregulated and downregulated genes respectively.
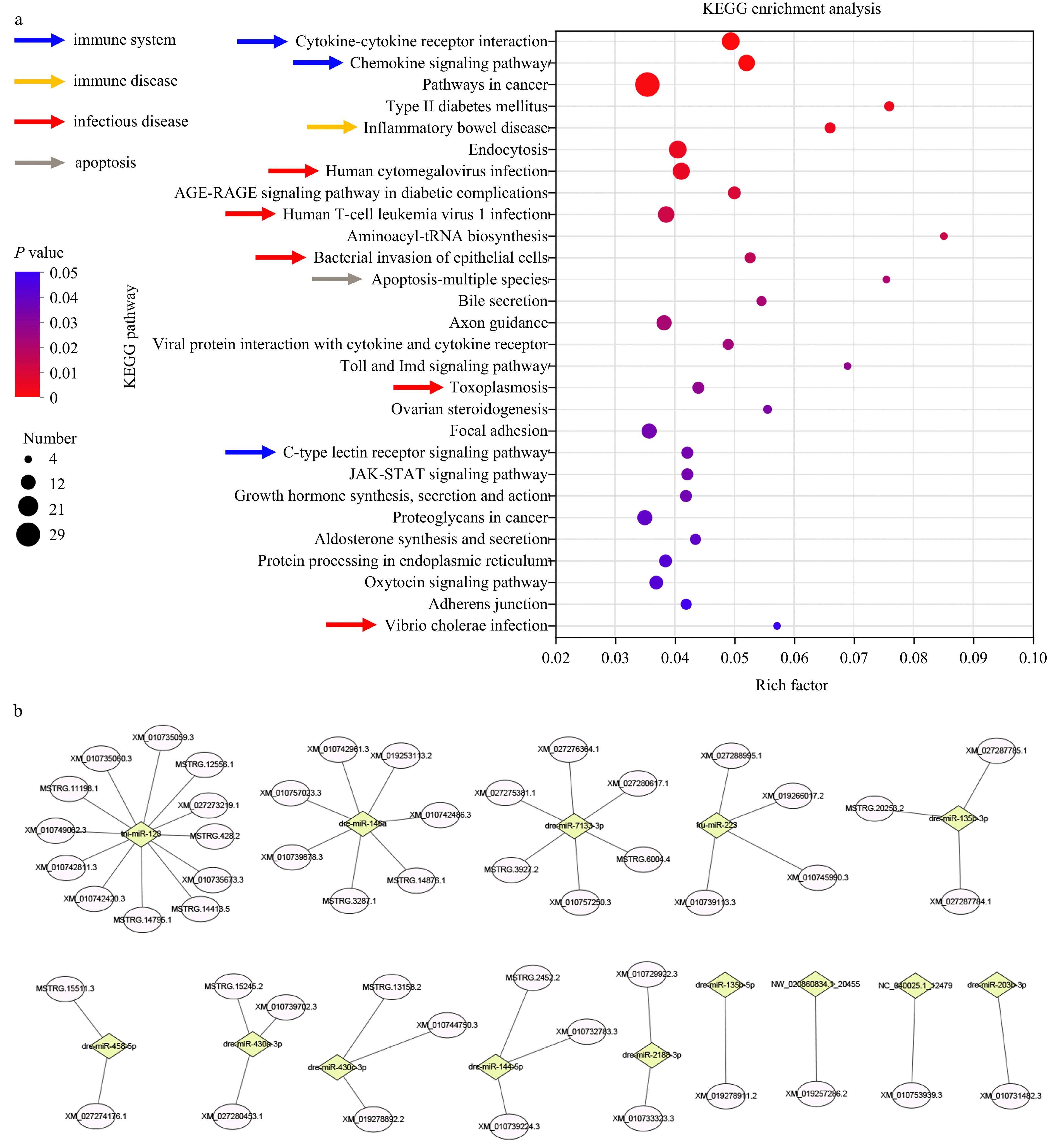
Figure 5. KEGG enrichment analysis of the miRNA targets and prediction of immune-related miRNA-mRNA pairs. a. KEGG enrichment analysis of the miRNA targets. Pathways involved in immune system, infectious and immune disease were indicated by arrows in different color. b. The immune-related miRNA-mRNA pairs. Diamond and ellipse represent miRNA and mRNA respectively.
-
Abo-Al-Ela H G. 2021. The emerging regulatory roles of noncoding RNAs in immune function of fish: MicroRNAs versus long noncoding RNAs. Molecular Genetics and Genomics, 296(4): 765–781, doi: 10.1007/s00438-021-01786-x Akayli T, Çanak Ö, Başaran B. 2011. A new pseudomonas species observed in cultured young rainbow trout (Oncorhynchus mykis Walbaum, 1792): Pseudomonas plecoglossicida. Biyoloji Bilimleri Araştırma Dergisi, 4(1): 107–111 Andreassen R, Høyheim B. 2017. miRNAs associated with immune response in teleost fish. Developmental & Comparative Immunology, 75: 77–85, doi: 10.1016/j.dci.2017.02.023 Ashida H, Mimuro H, Ogawa M, et al. 2011. Cell death and infection: a double-edged sword for host and pathogen survival. Journal of Cell Biology, 195(6): 931–942, doi: 10.1083/jcb.201108081 Baek D, Villén J, Shin C, et al. 2008. The impact of microRNAs on protein output. Nature, 455(7209): 64–71, doi: 10.1038/nature07242 Behar S M, Briken V. 2019. Apoptosis inhibition by intracellular bacteria and its consequence on host immunity. Current Opinion in Immunology, 60: 103–110, doi: 10.1016/j.coi.2019.05.007 Best S M. 2008. Viral subversion of apoptotic enzymes: escape from death row. Annual Review of Microbiology, 62: 171–192, doi: 10.1146/annurev.micro.62.081307.163009 Bulieris P V, Shaikh N H, Freddolino P L, et al. 2017. Structure of FlgK reveals the divergence of the bacterial Hook-Filament Junction of Campylobacter. Scientific Reports, 7(1): 15743, doi: 10.1038/s41598-017-15837-0 Çetin G, Klafack S, Studencka-Turski M, et al. 2021. The ubiquitin-proteasome system in immune cells. Biomolecules, 11(1): 60, doi: 10.3390/biom11010060 Chen Huazhi, Zhang Yameng, Shao Guangming, et al. 2023. Comparative transcriptomics reveals the microRNA‐mediated immune response of large yellow croaker (Larimichthys crocea) to Pseudomonas plecoglossicida infection. Fishes, 8(1): 10, doi: 10.3390/fishes8010010 Dang Yunfei, Xu Xiaoyan, Shen Yubang, et al. 2016. Transcriptome analysis of the innate immunity-related complement system in spleen tissue of Ctenopharyngodon idella infected with Aeromonas hydrophila. PLoS One, 11(7): e0157413, doi: 10.1371/journal.pone.0157413 de Graaf N, van Helden M J G, Textoris-Taube K, et al. 2011. PA28 and the proteasome immunosubunits play a central and independent role in the production of MHC class I-binding peptides in vivo. European Journal of Immunology, 41(4): 926–935, doi: 10.1002/eji.201041040 Friedländer M R, Mackowiak S D, Li N, et al. 2012. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Research, 40(1): 37–52, doi: 10.1093/nar/gkr688 Fu Yuanshuai, Xu Zhe, Wen Bin, et al. 2020. Gonad-specific transcriptomes reveal differential expression of gene and miRNA between male and female of the discus fish (Symphysodon aequifasciatus). Frontiers in Physiology, 11: 754, doi: 10.3389/fphys.2020.00754 Griffith J W, Sokol C L, Luster A D. 2014. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annual Review of Immunology, 32: 659–702, doi: 10.1146/annurev-immunol-032713-120145 Griffiths-Jones S, Grocock R J, van Dongen S, et al. 2006. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Research, 34(S1): D140–D144, doi: 10.1093/nar/gkj112 Huang Lixing, Liu Wenjia, Jiang Qingling, et al. 2018a. Integration of transcriptomic and proteomic approaches reveals the temperature-dependent virulence of Pseudomonas plecoglossicida. Frontiers in Cellular and Infection Microbiology, 8: 207, doi: 10.3389/fcimb.2018.00207 Huang Lixing, Zhao Lingmin, Su Yongquan, et al. 2018b. Genome sequence of Pseudomonas plecoglossicida strain NZBD9. Genome Announcements, 6(4): e01412–17, doi: 10.1128/genomeA.01412-17 Huang Lixing, Zuo Yanfei, Jiang Qingling, et al. 2019. A metabolomic investigation into the temperature-dependent virulence of Pseudomonas plecoglossicida from large yellow croaker (Pseudosciaena crocea). Journal of Fish Diseases, 42(3): 431–446, doi: 10.1111/jfd.12957 Jiao Jiping, Zhao Lingmin, Huang Lixing, et al. 2021. The contributions of fliG gene to the pathogenicity of Pseudomonas plecoglossicida and pathogen-host interactions with Epinephelus coioides. Fish & Shellfish Immunology, 119: 238–248, doi: 10.1016/j.fsi.2021.09.032 Kalvari I, Nawrocki E P, Argasinska J, et al. 2018. Non-coding RNA analysis using the rfam database. Current Protocols in Bioinformatics, 62(1): e51, doi: 10.1002/cpbi.51 Kalvari I, Nawrocki E P, Ontiveros-Palacios N, et al. 2021. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Research, 49(D1): D192–D200, doi: 10.1093/nar/gkaa1047 Kammerl I E, Meiners S. 2016. Proteasome function shapes innate and adaptive immune responses. American Journal of Physiology-Lung Cellular and Molecular Physiology, 311(2): L328–L336, doi: 10.1152/ajplung.00156.2016 Kanehisa M, Furumichi M, Tanabe M, et al. 2017. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Research, 45(D1): D353–D361, doi: 10.1093/nar/gkw1092 Khalil B A, Elemam N M, Maghazachi A A. 2021. Chemokines and chemokine receptors during COVID-19 infection. Computational and Structural Biotechnology Journal, 19: 976–988, doi: 10.1016/j.csbj.2021.01.034 Kim D, Paggi J M, Park C, et al. 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology, 37(8): 907–915, doi: 10.1038/s41587-019-0201-4 Klopfenstein D V, Zhang Liangsheng, Pedersen B S, et al. 2018. GOATOOLS: a Python library for Gene Ontology analyses. Scientific Reports, 8(1): 10872, doi: 10.1038/s41598-018-28948-z Kominami K, Nakabayashi J, Nagai T, et al. 2012. The molecular mechanism of apoptosis upon caspase-8 activation: quantitative experimental validation of a mathematical model. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 1823(10): 1825–1840, doi: 10.1016/j.bbamcr.2012.07.003 Krishnan R, Kim J O, Jang Y S, et al. 2021. Proteasome subunit beta type-8 from sevenband grouper negatively regulates cytokine responses by interfering NF-κB signaling upon nervous necrosis viral infection. Fish & Shellfish Immunology, 113: 118–124 Krüger J, Rehmsmeier M. 2006. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Research, 34(S2): W451–W454, doi: 10.1093/nar/gkl243 Labbé K, Saleh M. 2008. Cell death in the host response to infection. Cell Death & Differentiation, 15(9): 1339–1349, doi: 10.1038/cdd.2008.91 Langmead B, Trapnell C, Pop M, et al. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology, 10(3): R25, doi: 10.1186/gb-2009-10-3-r25 Lewis B P, Burge C B, Bartel D P. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell, 120(1): 15–20, doi: 10.1016/j.cell.2004.12.035 Li Bo, Dewey C N. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12(1): 323, doi: 10.1186/1471-2105-12-323 Li Wenrui, Guan Xiaolu, Jiang Shuai, et al. 2020a. The novel fish miRNA pol-miR-novel_171 and its target gene FAM49B play a critical role in apoptosis and bacterial infection. Developmental & Comparative Immunology, 106: 103616, doi: 10.1016/j.dci.2020.103616 Li Wenrui, Guan Xiaolu, Sun Li. 2020b. Phosphatase and tensin homolog (PTEN) of Japanese flounder-its regulation by miRNA and role in autophagy, apoptosis and pathogen infection. International Journal of Molecular Sciences, 21(20): 7725, doi: 10.3390/ijms21207725 Li Lu, Tong An, Zhang Qiangsheng, et al. 2021. The molecular mechanisms of MLKL-dependent and MLKL-independent necrosis. Journal of Molecular Cell Biology, 13(1): 3–14, doi: 10.1093/jmcb/mjaa055 Li Chengwei, Wang Shenglan, Ren Qiulei, et al. 2020c. An outbreak of visceral white nodules disease caused by Pseudomonas plecoglossicida at a water temperature of 12°C in cultured large yellow croaker (Larimichthys crocea) in China. Journal of Fish Diseases, 43(11): 1353–1361, doi: 10.1111/jfd.13206 Liu Hua, Xie Jiafang, Yu Hui, et al. 2021. The early response expression profiles of miRNA-mRNA in farmed yellow catfish (Pelteobagrus fulvidraco) challenged with Edwardsiella tarda infection. Developmental & Comparative Immunology, 119: 104018, doi: 10.1016/j.dci.2021.104018 Liu Zixu, Zhao Lingmin, Huang Lixing, et al. 2020. Integration of RNA-seq and RNAi provides a novel insight into the immune responses of Epinephelus coioides to the impB gene of Pseudomonas plecoglossicida. Fish & Shellfish Immunology, 105: 135–143, doi: 10.1016/j.fsi.2020.06.023 Love M I, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 15(12): 550, doi: 10.1186/s13059-014-0550-8 Luo Gang, Xu Xiaojin, Zhao Lingmin, et al. 2019. clpV is a key virulence gene during in vivo Pseudomonas plecoglossicida infection. Journal of Fish Diseases, 42(7): 991–1000, doi: 10.1111/jfd.13001 Maekawa S, Wang Peichi, Chen Shin-Chu. 2019. Comparative study of immune reaction against bacterial infection from transcriptome analysis. Frontiers in Immunology, 10: 153, doi: 10.3389/fimmu.2019.00153 Mao Zhijuan, Li Meifang, Chen Jigang. 2013. Draft genome sequence of Pseudomonas plecoglossicida Strain NB2011, the causative agent of white nodules in large yellow croaker (Larimichthys crocea). Genome Announcements, 1(4): e00586–13, doi: 10.1128/genomea.00586-13 Martin C J., Booty M G, Rosebrock T R, et al. 2012. Efferocytosis is an innate antibacterial mechanism. Cell Host & Microbe, 12(3): 289–300, doi: 10.1016/j.chom.2012.06.010 McIlwain D R, Berger T, Mak T W. 2013. Caspase functions in cell death and disease. Cold Spring Harbor Perspectives in Biology, 5(4): a008656, doi: 10.1101/cshperspect.a008656 Meng Xianmei, Dang Tong, Chai Jianyuan. 2021. From apoptosis to necroptosis: the death wishes to cancer. Cancer Control, 28: 10732748211066311, doi: 10.1177/10732748211066311 Ministry of Agriculture and Rural Affairs, National Fisheries Technology Extension Center, China Society of Fisheries. 2024. China Fishery Statistical Yearbook (in Chinese). Beijing: China Agriculture Press, 117 Mootha V K, Lindgren C M, Eriksson K F, et al. 2003. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genetics, 34(3): 267–273, doi: 10.1038/ng1180 Murata S, Takahama Y, Kasahara M, et al. 2018. The immunoproteasome and thymoproteasome: functions, evolution and human disease. Nature Immunology, 19(9): 923–931, doi: 10.1038/s41590-018-0186-z Nailwal H, Chan F K M. 2019. Necroptosis in anti-viral inflammation. Cell Death & Differentiation, 26(1): 4–13, doi: 10.1038/s41418-018-0172-x Ni Songwei, Yan Yang, Cui Huachun, et al. 2017. Fish miR-146a promotes Singapore grouper iridovirus infection by regulating cell apoptosis and NF-κB activation. The Journal of General Virology, 98(6): 1489–1499, doi: 10.1099/jgv.0.000811 Nishimori E, Kita-Tsukamoto K, Wakabayashi H. 2000. Pseudomonas plecoglossicida sp. nov. , the causative agent of bacterial haemorrhagic ascites of ayu, Plecoglossus altivelis. International Journal of Systematic and Evolutionary Microbiology, 50(1): 83–89, doi: 10.1099/00207713-50-1-83 Nogusa S, Thapa R J, Dillon C P, et al. 2016. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host & Microbe, 20(1): 13–24, doi: 10.1016/j.chom.2016.05.011 Oo Y H, Shetty S, Adams D H. 2010. The role of chemokines in the recruitment of lymphocytes to the liver. Digestive Diseases, 28(1): 31–44, doi: 10.1159/000282062 Ou Jiangtao, Chen Hao, Liu Qiao, et al. 2021. Integrated transcriptome analysis of immune-related mRNAs and microRNAs in Macrobrachium rosenbergii infected with Spiroplasma eriocheiris. Fish & Shellfish Immunology, 119: 651–669, doi: 10.1016/j.fsi.2021.11.002 Pertea M, Pertea G M, Antonescu C M, et al. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology, 33(3): 290–295, doi: 10.1038/nbt.3122 Powers R K, Goodspeed A, Pielke-Lombardo H, et al. 2018. GSEA-InContext: identifying novel and common patterns in expression experiments. Bioinformatics, 34(13): i555–i564, doi: 10.1093/bioinformatics/bty271 Qian Youcun, Kang Zizhen, Liu Caini, et al. 2010. IL-17 signaling in host defense and inflammatory diseases. Cellular & Molecular Immunology, 7(5): 328–333, doi: 10.1038/cmi.2010.27 Riffo-Campos Á L, Riquelme I, Brebi-Mieville P. 2016. Tools for sequence-based miRNA target prediction: what to choose? International Journal of Molecular Sciences, 17(12): 1987, doi: 10.3390/ijms17121987 Rosenblatt C K, Harriss A, Babul A N, et al. 2021. Machine learning for subtyping concussion using a clustering approach. Frontiers in Human Neuroscience, 15: 716643, doi: 10.3389/fnhum.2021.716643 Shannon P, Markiel A, Ozier O, et al. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research, 13(11): 2498–2504, doi: 10.1101/gr.1239303 Shen Yawei, Zhao Ziwei, Zhao Jinliang, et al. 2019. Expression and functional analysis of hepcidin from mandarin fish (Siniperca chuatsi). International Journal of Molecular Sciences, 20(22): 5602, doi: 10.3390/ijms20225602 Son K, Yu S, Shin W, et al. 2018. A simple guideline to assess the characteristics of RNA-Seq data. BioMed Research International, 2018: 2906292, doi: 10.1155/2018/2906292 Sun Yujia, Nie Pin, Zhao Lingmin, et al. 2019a. Dual RNA-Seq unveils the role of the Pseudomonas plecoglossicida fliA gene in pathogen-host interaction with Larimichthys crocea. Microorganisms, 7(10): 443, doi: 10.3390/microorganisms7100443 Sun Yujia, Zhuang Zhixia, Wang Xiaoru, et al. 2019b. Dual RNA-seq reveals the effect of the flgM gene of Pseudomonas plecoglossicida on the immune response of Epinephelus coioides. Fish & Shellfish Immunology, 87: 515–523, doi: 10.1016/j.fsi.2019.01.041 Sundaray J K, Dixit S, Rather A, et al. 2022. Aquaculture omics: an update on the current status of research and data analysis. Marine Genomics, 64: 100967, doi: 10.1016/j.margen.2022.100967 Tanzer M C. 2022. A proteomic perspective on TNF-mediated signalling and cell death. Biochemical Society Transactions, 50(1): 13–20, doi: 10.1042/BST20211114 Tian Xue, Pang Xiaolei, Wang Liangyan, et al. 2018. Dynamic regulation of mRNA and miRNA associated with the developmental stages of skin pigmentation in Japanese ornamental carp. Gene, 666: 32–43, doi: 10.1016/j.gene.2018.04.054 Wang Luying, Sun Yunjia, Zhao Lingmin, et al. 2019. Dual RNA-seq uncovers the immune response of Larimichthys crocea to the secY gene of Pseudomonas plecoglossicida from the perspective of host-pathogen interactions. Fish & Shellfish Immunology, 93: 949–957, doi: 10.1016/j.fsi.2019.08.040 Wang Liguo, Wang Shengqin, Li Wei. 2012. RSeQC: quality control of RNA-seq experiments. Bioinformatics, 28(16): 2184–2185, doi: 10.1093/bioinformatics/bts356 Wang Longlong, Yu Fei, Xu Ning, et al. 2020. Grass carp reovirus capsid protein interacts with cellular proteasome subunit beta-type 7: evidence for the involvement of host proteasome during aquareovirus infection. Fish & Shellfish Immunology, 98: 77–86, doi: 10.1016/j.fsi.2019.12.047 Wu Ronghua, Sheng Xiuzhen, Tang Xiaoqian, et al. 2018. Transcriptome analysis of flounder (Paralichthys olivaceus) gill in response to lymphocystis disease virus (LCDV) infection: novel insights into fish defense mechanisms. International Journal of Molecular Sciences, 19(1): 160, doi: 10.3390/ijms19010160 Zhang Yameng, Lu Lixia, Li Chengwei, et al. 2021a. Transcriptome analysis revealed multiple immune processes and energy metabolism pathways involved in the defense response of the large yellow croaker Larimichthys crocea against Pseudomonas plecoglossicida. Comparative Biochemistry and Physiology Part D: Genomics and Proteomics, 40: 100886, doi: 10.1016/j.cbd.2021.100886 Zhang Beibei, Luo Gang, Zhao Lingmin, et al. 2018. Integration of RNAi and RNA-seq uncovers the immune responses of Epinephelus coioides to L321_RS19110 gene of Pseudomonas plecoglossicida. Fish & Shellfish Immunology, 81: 121–129, doi: 10.1016/j.fsi.2018.06.051 Zhang Chi, Tu Jiagang, Zhang Yongan. 2021b. MicroRNA regulation of viral replication in teleost fish: a review. Reviews in Aquaculture, 13(3): 1367–1378, doi: 10.1111/raq.12526 Zhang J T, Zhou S M, An S W, et al. 2014. Visceral granulomas in farmed large yellow croaker, Larimichthys crocea (Richardson), caused by a bacterial pathogen, Pseudomonas plecoglossicida. Journal of fish diseases, 37(2): 113–121, doi: 10.1111/jfd.12075 Zhou Wei, Xie Yadong, Li Yu, et al. 2021. Research progress on the regulation of nutrition and immunity by microRNAs in fish. Fish & Shellfish Immunology, 113: 1–8, doi: 10.1016/j.fsi.2021.03.011 -
 DownLoad:
DownLoad:
 下载:
下载:
 点击查看大图
点击查看大图
计量
- 文章访问数: 14
- HTML全文浏览量: 5
- 被引次数: 0

